- 移动端


上海优宁维生物科技股份有限公司代理商
19 年
手机商铺
- NaN
- 0
- 0
- 2
- 2


斯达特
抗体/ELISA 试剂盒/试剂
已认证品牌介绍

Absin
生物化学/抗体
已认证品牌介绍

LabEx
技术服务
已认证品牌介绍

Cytiva
实验室仪器 / 设备/试剂/耗材/细胞库 / 细胞培养
已认证品牌介绍

Novus
抗体/试剂
已认证品牌介绍

Sigma-Aldrich
实验室仪器 / 设备/试剂/耗材
已认证品牌介绍

Thermo Fisher
试剂/实验室仪器 / 设备/耗材
已认证品牌介绍

Horizon Discovery
细胞库 / 细胞培养/技术服务/动物模型
已认证品牌介绍

Enzo
实验室仪器 / 设备/试剂
已认证品牌介绍

BiosPacific
抗体
已认证品牌介绍

Cayman
抗体/试剂
已认证品牌介绍

Bio X Cell
抗体/试剂
已认证品牌介绍

R&D Systems
试剂
已认证品牌介绍

Tocris bioscience
试剂
已认证品牌介绍

Innova Biosciences
抗体
已认证品牌介绍

Cell Signaling Technology
抗体
已认证品牌介绍

Biovision
试剂
已认证品牌介绍

Fitzgerald
试剂
已认证品牌介绍

LifeSpan BioSciences
抗体/试剂
已认证品牌介绍

AnaSpec
试剂
已认证品牌介绍

Sengenics
试剂/技术服务
已认证品牌介绍

Cytoskeleton
蛋白质/抗原/多肽/试剂
已认证品牌介绍

Synaptic Systems
试剂
已认证品牌介绍

USBiological
抗体/试剂
已认证品牌介绍

Agrisera
抗体
已认证品牌介绍

Meridian
抗体/试剂
已认证品牌介绍

Miltenyi Biotec
实验室仪器 / 设备/试剂
已认证品牌介绍

Alomone Labs
试剂
已认证品牌介绍
斯达特
抗体/ELISA 试剂盒/试剂
已认证品牌介绍
Absin
生物化学/抗体
已认证品牌介绍
LabEx
技术服务
已认证品牌介绍
Cytiva
实验室仪器 / 设备/试剂/耗材/细胞库 / 细胞培养
已认证品牌介绍
推荐产品




技术资料/正文
Nature三连,非编码DNA成癌症研究新热点
368 人阅读发布时间:2020-12-10 14:06
RNA剪接在基因表达中占据核心位置,其主要参与基因转录过程中,内含子切除、外显子拼接的一步。正确的RNA剪接会形成正确的mRNA,反之,则会使原本序列中新增一段非编码DNA序列,生成错误的mRNA,进而形成错误的蛋白质,导致疾病。
最新的研究发现,RNA的异常剪接与U2 snRNP的剪接因子SF3B1的突变相关,而SF3B1是最常被检测到的突变的RNA剪接因子。据报道,在髓系白血病、淋巴系白血病和实体瘤中,SF3B1在特定残基位点处反复发生错误突变。RNA剪接过程主要由U1-U6完成,其中U1主要功能是识别5’剪接位点;U2主要功能是识别3’剪接位点,突变的SF3B1则导致异常3’剪接位点被识别。
2019年10月9日,《Nature》杂志同期刊登出三篇文章对癌症中剪接因子突变进行报道。接下来小优带您详细了解一下:
SF3B1驱动黑色素瘤的进展

[1] Daichi Inoue et al. Spliceosomal disruption of the non-canonical BAF complex in cancer, Nature.
DOI: https://doi.org/10.1038/s41586-019-1646-9
第一篇来自于Omar Abdel-Wahab和Robert K. Bradley的合作研究,利用全癌剪接分析和CRISPR技术,鉴定得出不同的SF3B1突变都会抑制BRD9,BRD9是非典型BAF(ncBAF)染色体重塑复合物的核心组分,是一种重要的抑癌蛋白。突变的SF3B1识别BRD9基因内一个异常的内含子分支点,从而诱导内源性逆转录病毒元件的毒性外显子的形成,致BRD9的mRNA发生降解。BRD9的缺失则会导致CTCF相关基因位点上ncBAF的丢失,促进黑色素瘤的发生,故在SF3B1突变细胞中纠正BRD9的错误剪切能够有效抑制肿瘤生长。
研究人员对SF3B1突变细胞系和多数慢性淋巴系白血病、骨髓增生异常综合征和UVM患者样本的错误剪接事件进行了分析,然后利用CRISPR阳性富集筛选系统进行功能分析。结果显示:BRD9的缺失促进细胞转化,且在所有携带SF3B1突变的癌症患者的BRD9基因中,均存在RNA的错误剪接现象;对该基因进行敲除后,人类UVM、皮肤黑色素瘤、胰腺癌细胞就会生长;SF3B1突变导致BRD9内含子序列的外显子化,即毒性外显子形成,该变化可介导RNA降解。
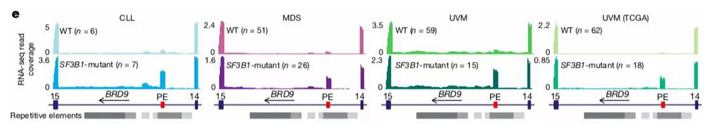
SF3B1突变导致BRD9毒性外显子形成
利用CRISPR-Cas9技术对BRD9毒性外显子进行修正后,该外显子中的夹杂物得到删除修正。
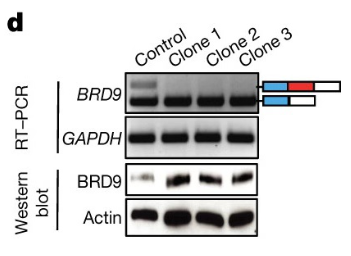
基因编辑后的BRD9毒性夹杂物丢失
BRD9与GLTSCR1是一部分ncBAF的组成物,SF3B1突变会使BRD9基因缺失。在UVM中,BRD9的丢失会优先影响参与凋亡与细胞生长、粘附和迁移的基因。BRD9降解后,BRD9和BRG1、GLTSCR1之间的相互作用力也随之消失,但BRG1和BAF155的相互作用仍保持完整,说明SF3B1突变体特异性破坏了ncBAF。
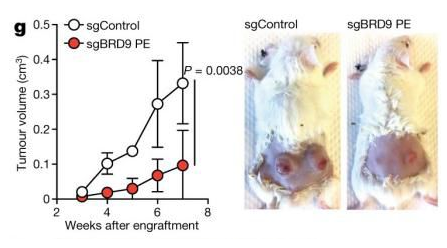
SF3B1突变细胞中的BRD9剪接影响肿瘤生长
那么BRD9缺失后,基因表达会出现什么变化呢?研究者对具有启动子的基因进行了研究,结果显示:在这些启动子中,BRD9可以跟ncBAF进行结合,且能够在突变型与野生型SF3B1的UVM患者中进行差异表达。ncBAF结合力的丧失与基因表达的提升和抑制有关,足以表明ncBAF既具有激活作用又具有抑制作用。
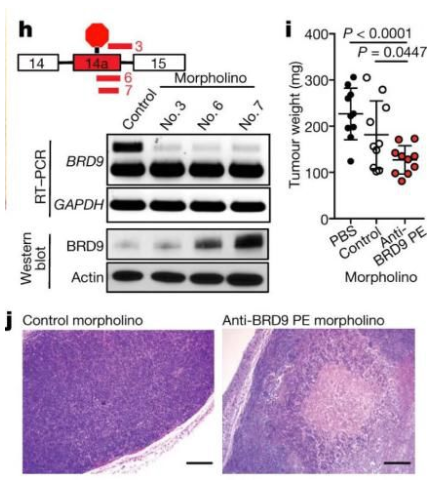
BRD9是黑色素瘤的可治疗靶向肿瘤抑制因子
接下来,小优对此篇文章里的实验思路及所用到的产品做了如下整理(实验中涉及到的产品小优都可以提供哦):
1
文献思路
将黑色素瘤细胞系移植于SCID小鼠(Jackson Laboratories)体内,将慢性淋巴细胞白血病患者的细胞系移植于NSG小鼠(Jackson Laboratories)体内,每天对小鼠进行严格的疾病或发病迹象监测。在小鼠体内发现肿瘤体积超过1cm3,小鼠出现明显的体重减轻、皮肤病、出血以及感染现象。
使用优化的CRISPR设计工具(http://crispr.mit.edu)选择出靶向BRD9转录起始位点的guide RNA序列,用同源重组法将其克隆至px458-GFP载体中。由于该序列含有HA标签,故其与HA标签上游的BRD9 5'UTR具有同源性。使用Lonza Nucleofector V试剂盒将靶向构建体转染至K562 SF3B1K700E细胞和MEL270细胞中,随后进行细胞分选。将SF3B1 cDNA在K562细胞中进行过表达,通过RNeasy Mini or Micro kit (Qiagen)分离得出总RNA,再使用uperScript VILO cDNA synthesis kit (Life Technologies)将其转录成cDNA,稀释后使用SYBR Green PCR Master Mix进行定量RT-PCR分析,上述分析在Applied Biosystems QuantStudio 6 Flex循环仪 (ThermoFisher Scientific)上进行。
2
细胞培养
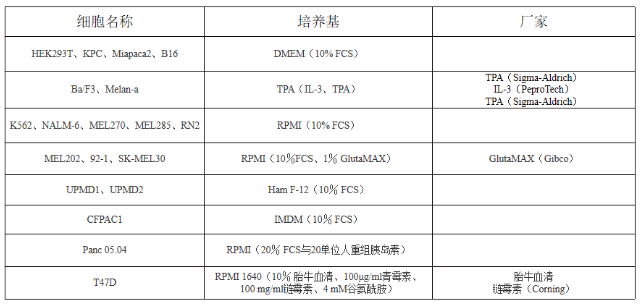
以上所有细胞培养基均含青霉素和链霉素
3
WB实验
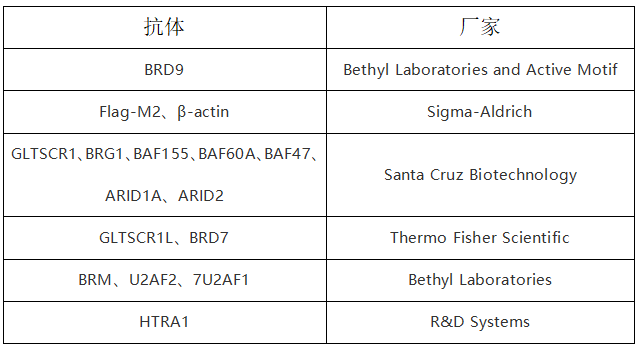
将一抗用5% BSA(Sigma-Aldrich)稀释,取2–5μg抗体与protein A/G PLUS Agarose于4°C下孵育3小时(Santa Cruz Biotechnology),通过Pierce IP裂解缓冲(Thermo Fisher Scientific)洗涤3次后,用Pierce Lane Marker Reducing Sample Buffer(Thermo Fisher Scientific)洗脱免疫沉淀的蛋白,然后上样至4–12%Bis-Tris NuPAGE Gels(Life Technologies)。
4
ChIP实验
抗体:BRG1、BRD9 (Abcam)
用空载体对MEL270细胞进行转导,用强力霉素或DMSO处理野生型SF3B1 cDNA和SF3B1K700E cDNA。
5
质谱检测实验
将用空载体或带Flag标签的BRD9转导的K562细胞于含10% FCS的RPMI中培养,随后进行质谱分析。细胞用1%不含甲醇的甲醛(Sigma)固定8分钟,并用0.125 M甘氨酸(Sigma)进行淬灭。通过添加裂解缓冲液分离染色质,然后用Dounce匀浆器破碎。对裂解液进行超声处理后,将DNA剪切至300–500 bp。通过用RNase、蛋白酶K和加热处理染色质等方式对其进行反向交联,然后用乙醇沉淀获取gDNA。将球粒重悬,并将所得的gDNA在NanoDrop分光光度计上进行定量。用Protein G(Invitrogen)预清洗等分的染色质,并对所需蛋白进行免疫沉淀。洗涤蛋白质复合物,去除免疫沉淀物和蛋白质消化物。将蛋白质消化物与磁珠分离,使用C18离心柱(Harvard Apparatus)进行纯化。使用SpeedVac将肽段进行真空干燥。在Thermo Scientific Q Exactive Orbitrap质谱仪上,采用Proxeon Easy-nLC II HPLC(Thermo Scientific)和Proxeon nanospray source,通过液相色谱和串联质谱法对消化的肽段进行分析。
6
shRNA实验
用强力霉素诱导的LT3GEPIR慢病毒载体T3G-GFP-mirE-PGK-Puro-IRES-rtTA334转导细胞, 表达针对BRD9的shRNA或针对雷尼拉的非靶向shRNA。加入强力霉素(Sigma Aldrich)诱导shRNA。
7
siRNA转染实验
用非靶向对照siRNA(Dharmacon),针对U2AF1的siRNA库(Dharmacon)或针对U2AF2的siRNA库转染K562细胞 (Dharmacon),使用Lonza的Nucleofector II设备和Cell Line Nucleofector Kit V对其进行转染和RNA、蛋白质的提取。使用1μg RNA和Superscript III第一链合成系统(Thermo Fisher)生成cDNA。
为分析sgRNA对BRD9的作用,先用LentiCas9-Blast(Addgene)转导细胞系,然后将其单细胞分选到96孔板中。将gRNA序列克隆至iLenti-guide-GFP载体中,对表达Cas9的细胞进行慢病毒转导,发现sgRNA的表达与GFP的表达有关。然后使用BD LSRFortessa测量表达GFP的细胞的百分比,计算GFP的阳性率。同样,为了分析cDNA、HTRA1 cDNA或shRNA对BRD9或HTRA1的作用,分别将pMIGII-主链质粒(cDNA)和LT3GEPIR质粒(shRNA)导入黑素瘤细胞后,计算GFP阳性率。
8
CRISPR富集筛选
用LentiCas9-Blast(Addgene)转导Ba/F3细胞,并将单细胞分选到96孔板中。在这些克隆中,使用了具有强Cas9表达的单克隆。SF3B1突变细胞中NMD靶标的sgRNA文库被扩增并转导为慢病毒。该文库包含针对每个靶基因的4个sgRNA,针对Pten的100个对照sgRNA和阳性对照sgRNA。用293FT细胞产生的携带慢病毒的sgRNA文库转导Ba/F3细胞,并在含IL-3的培养基中进行嘌呤霉素选择。洗出IL-3,并在IL-3耗尽后收集了存活的细胞。裂解细胞沉淀,提取gDNA(Qiagen),并通过Qubit(Thermo Scientific)进行定量。使用Q5高保真聚合酶(NEB)对gDNA进行PCR扩增,添加Illumina衔接子和多重条形码。通过Qubit和Bioanalyzer(Agilent)对扩增子进行定量,并在Illumina HiSeq 2500上进行测序。对每个gRNA进行计数,并在细胞因子耗竭后第0天到第7天比较每个sgRNA数量。对于探针水平的分析,我们拟合了负二项式广义对数线性模型,并在Bioconductor edgeR软件包中使用glmFit和glmLRT进行了likelihood ratio测试。对于基因水平的分析,在edgeR36–38中进行CAMERA测试,使用Benjamini-Hochberg方法计算FDR值。
用靶向BRD9毒物外显子5'末端的iLenti-guide-Puro载体转导表达Cas9的MEL202和MEL270细胞。在选择嘌呤霉素后,使用BD FACSAria III细胞分选仪将单细胞分选到96孔板中。通过PCR扩增,然后进行Sanger测序,确认突变是由Cas9和sgRNA引起。
9
菌落鉴定
由MEL202和MEL270细胞制备单细胞悬液,在有或没有CRISPR介导的情况下,将来自细胞系的3000个细胞一式三份置于预处理的平板中,10天后对菌落进行计数。用3.7%多聚甲醛固定菌落5分钟,并在室温下于0.05%结晶紫溶液中染色30分钟,然后用PBS和自来水洗涤。
10
体外细胞活力测定
将细胞以每孔1000个细胞的密度接种在白色平孔96孔板中,每隔24小时使用Cell Titer Glo(Promega)进行发光读数。
11
BRD9基因测定
将Flag–SF3B1(WT)或Flag–SF3B1(K700E)转导至SF3B1野生型MEL270和T47D细胞中,用新霉素进行筛选(Thermo Fisher Scientific),随后用强力霉素(Sigma)处理所选细胞。使用Agilent QuikChange II site-directed mutagenesis kit对BRD9进行诱变,转染前,将细胞接种至24孔板中,转染后收集细胞,使用Qiagen RNeasy mini kit提取RNA,通过RT-PCR分析BRD9基因特性。
U1-snRNA突变驱动Shh髓母细胞瘤

[2] Hiromichi Suzuki et al. Recurrent non-coding U1-snRNA mutations drive cryptic splicing in Shh medulloblastoma, Nature.
DOI: https://doi.org/10.1038/s41586-019-1650-0.
第二篇,研究者发现近一半的Sonic hedgehog髓母细胞瘤的UI剪接因子的snRNA会产生高频热点突变,患病人群主要为成年人与青少年,且该突变不存在于其他髓母细胞瘤亚种中。突变主要发生在5'剪接位点结合区域,且snRNA突变型肿瘤会使RNA剪接发生错误,造成PTCH1蛋白失活,从而激活了GLI2,CCND2两种癌蛋白,其中PTCH1为一种抑癌因子。这项研究发现了髓母细胞瘤的治疗新靶点,并构成了癌症中非蛋白质编码基因的高度复发性和组织特异性突变。
U1剪接体RNA在多种癌症中反复突变

[3] Shimin Shuai et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers, Nature.
DOI: https://doi.org/10.1038/s41586-019-1651-z
第三篇,研究者收集了多种癌症样本,经分析发现在U1 snRNA的高频热点突变的第3个碱基上发生A>C的体细胞突变,从而使U1和5’剪接位点间的偏好性发生改变,原本的A-U配对变成了C-G配对,RNA剪接发生变化。在临床上,A> C突变与肝细胞癌(HCC)的酒精滥用和慢性淋巴细胞性白血病(CLL)的侵袭性IGHV亚型相关。该研究证明了剪接体RNA中最早的非编码驱动程序之一,揭示了癌症中异常剪接新机制,有望成为癌症新的治疗靶标。
总结
以上三篇同期发表在《Nature》的文章揭示了 RNA的错误剪接导致患者体内的抑癌因子失活,同时却激活了原癌基因,可谓是癌症“好助手”。这些异常剪接研究机制会在癌症治疗新靶标的发现中发挥重要意义。
RNA错误剪切与非编码DNA在肿瘤研究领域占据着相当重要的地位,未来很可能成为肿瘤治疗的新研究热点。有哪些技术可以应用到该领域的研究呢?NGS测序和基因编辑必不可少,相关产品推荐:
1.二代测序建库试剂盒

2.基因编辑产品
guide RNA产品
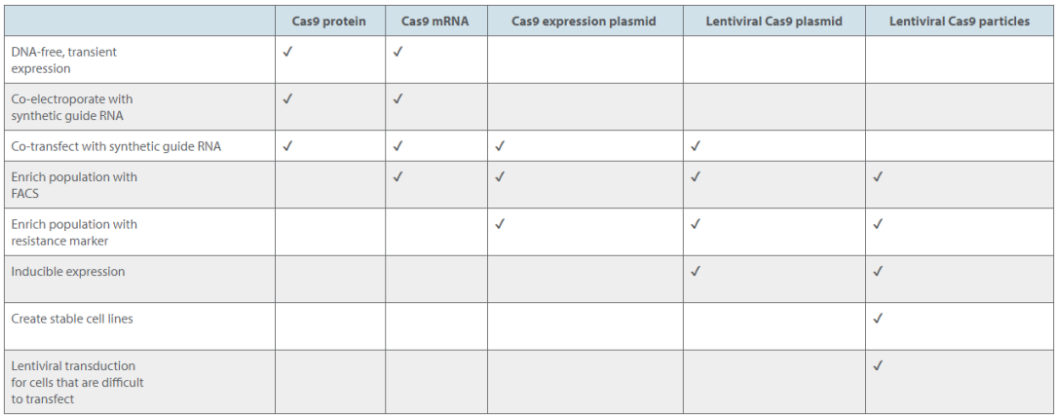
cas9产品
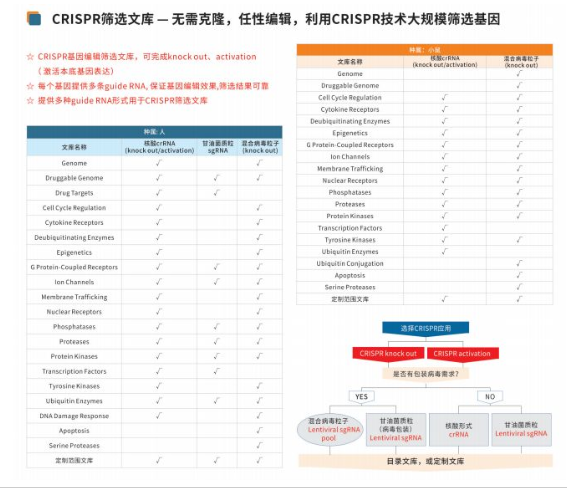
CRISPR筛选文库
【联系方式】
上海优宁维生物科技股份有限公司
抗体专家|流式专家|免疫组化|实验服务|科学仪器

免费热线:4008-168-068
咨询邮箱:info@univ-bio.com
官网:www.univ-bio.com
◁欢迎关注
长按二维码
点击关注~请多指教~