- 移动端


上海优宁维生物科技股份有限公司代理商
19 年
手机商铺
- NaN
- 0
- 0
- 2
- 2


斯达特
抗体/ELISA 试剂盒/试剂
已认证品牌介绍

Absin
生物化学/抗体
已认证品牌介绍

LabEx
技术服务
已认证品牌介绍

Cytiva
实验室仪器 / 设备/试剂/耗材/细胞库 / 细胞培养
已认证品牌介绍

Novus
抗体/试剂
已认证品牌介绍

Sigma-Aldrich
实验室仪器 / 设备/试剂/耗材
已认证品牌介绍

Thermo Fisher
试剂/实验室仪器 / 设备/耗材
已认证品牌介绍

Horizon Discovery
细胞库 / 细胞培养/技术服务/动物模型
已认证品牌介绍

Enzo
实验室仪器 / 设备/试剂
已认证品牌介绍

BiosPacific
抗体
已认证品牌介绍

Cayman
抗体/试剂
已认证品牌介绍

Bio X Cell
抗体/试剂
已认证品牌介绍

R&D Systems
试剂
已认证品牌介绍

Tocris bioscience
试剂
已认证品牌介绍

Innova Biosciences
抗体
已认证品牌介绍

Cell Signaling Technology
抗体
已认证品牌介绍

Biovision
试剂
已认证品牌介绍

Fitzgerald
试剂
已认证品牌介绍

LifeSpan BioSciences
抗体/试剂
已认证品牌介绍

AnaSpec
试剂
已认证品牌介绍

Sengenics
试剂/技术服务
已认证品牌介绍

Cytoskeleton
蛋白质/抗原/多肽/试剂
已认证品牌介绍

Synaptic Systems
试剂
已认证品牌介绍

USBiological
抗体/试剂
已认证品牌介绍

Agrisera
抗体
已认证品牌介绍

Meridian
抗体/试剂
已认证品牌介绍

Miltenyi Biotec
实验室仪器 / 设备/试剂
已认证品牌介绍

Alomone Labs
试剂
已认证品牌介绍
斯达特
抗体/ELISA 试剂盒/试剂
已认证品牌介绍
Absin
生物化学/抗体
已认证品牌介绍
LabEx
技术服务
已认证品牌介绍
Cytiva
实验室仪器 / 设备/试剂/耗材/细胞库 / 细胞培养
已认证品牌介绍
推荐产品




技术资料/正文
文献解析|CD36对动脉粥样硬化形成的作用机制
1496 人阅读发布时间:2025-02-18 14:53
CD36
前 言
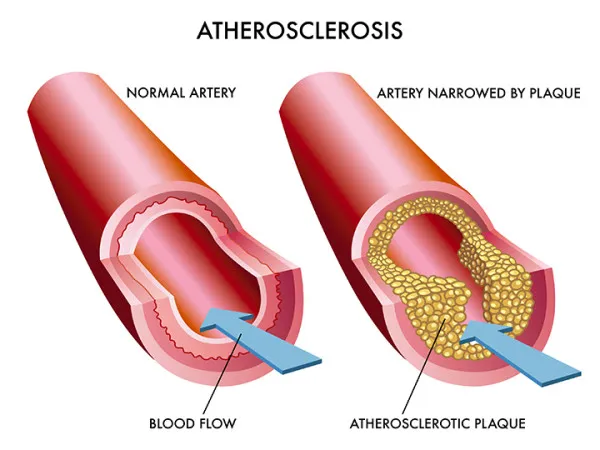
巨噬细胞和血管平滑肌细胞(VSMCs)具备多种清道夫受体的表达能力,这些受体能够捕获并内化来自血管内皮下层空间经过氧化修饰的低密度脂蛋白(ox-LDL)。这一过程促使这些细胞转化为泡沫细胞,为动脉粥样硬化斑块的形成提供了助力。随着斑块的不断演进和稳定性下降,可能会引发一系列临床症状和急性心血管事件。值得注意的是,CD36作为一种关键的B类清道夫受体,在巨噬细胞摄取ox-LDL的过程中占据了主导地位,其介导的摄取量高达60%-70%。

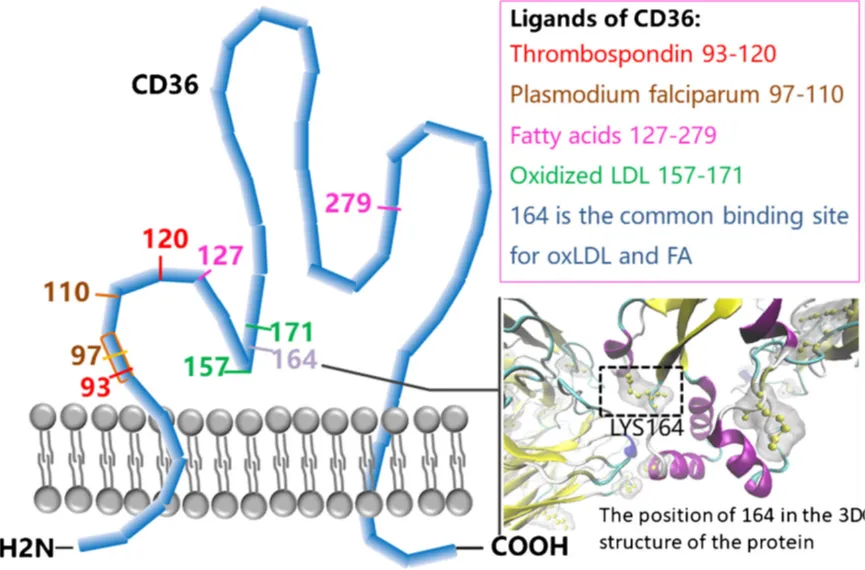
CD36的结构
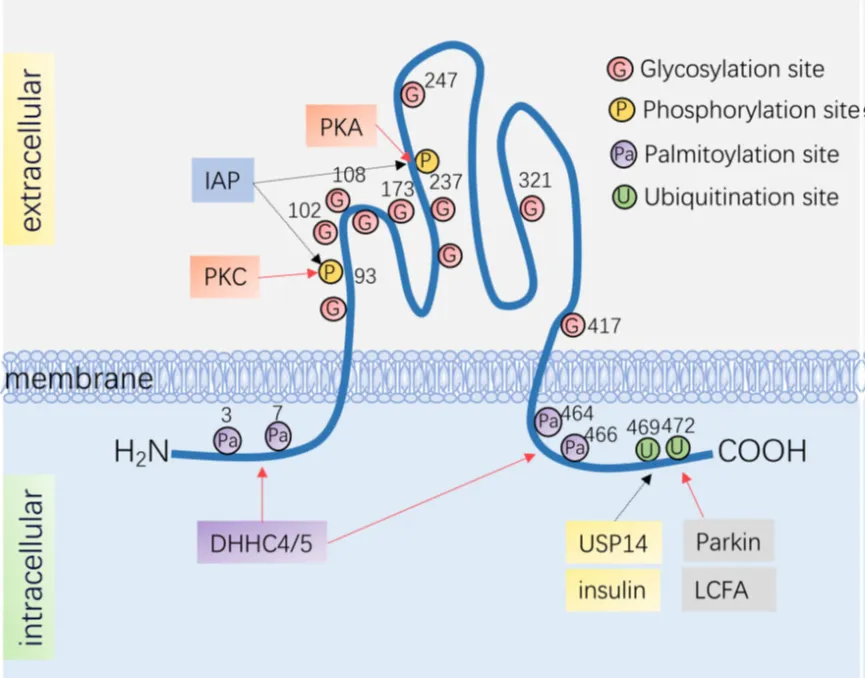
CD36是一种分子量约为88千道尔顿(kDa)的跨膜糖蛋白受体,归类于B类清道夫受体家族的一员。其基因定位于7号染色体的q11.2区域,结构由17个外显子和相邻的18个内含子组成。CD36的蛋白质结构由472个氨基酸残基构成,这些氨基酸折叠成一条单肽链,形成了两个穿越细胞膜的结构域,以及两个位于细胞质内的极短结构域。此外,它还包含一个庞大的糖基化胞外结构域,整体呈现出一种发夹状的膜拓扑结构特征。
CD36蛋白的翻译后修饰机制与功能影响
CD36的合成量、分布模式及其功能特性深受多种转录后修饰方式的调控,这些修饰包括泛素化、糖基化、磷酸化及棕榈酰化等过程。尽管CD36上存在多个潜在的乙酰化位点,但其具体作用机制仍有待深入探索。在CD36的N端区域,存在两个关键的泛素化位点,即Lys472和Lys469。当环境中存在长链脂肪酸时,CD36的泛素化水平会有所提升。值得注意的是,Parkin作为一种E3泛素连接酶,能够催化CD36的单泛素化,进而增强其稳定性;而USP14则负责介导CD36的去泛素化过程。
在CD36的C端和N端,还分布着四个棕榈酰化位点,分别是Cys3、Cys7、Cys464和Cys466。这些位点的棕榈酰化过程由DHHC4/5棕榈酰转移酶所催化。此外,CD36还拥有两个磷酸化位点,即Thr93和Ser237,它们可以分别被蛋白激酶C(PKC)、蛋白激酶A(PKA)以及IAP所磷酸化。
在CD36的细胞外结构域中,还发现了十个糖基化位点。这些翻译后修饰方式共同作用于CD36,对其功能特性产生深远的影响。
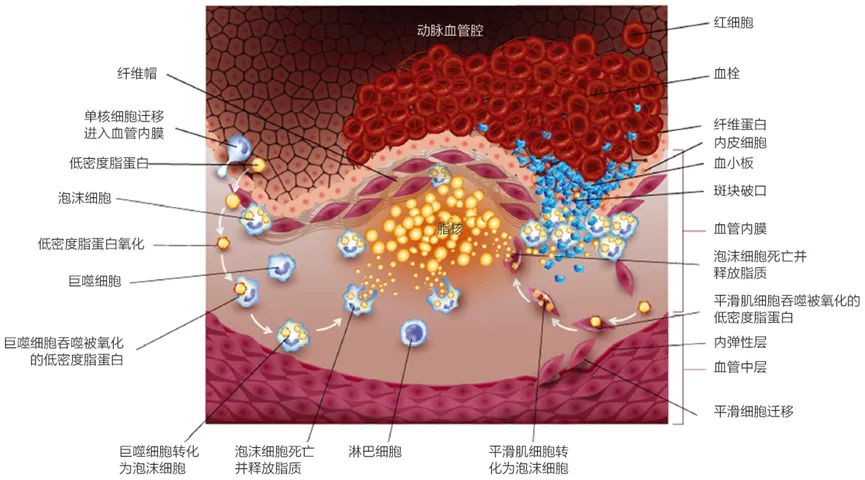
CD36在内皮功能障碍中的作用机制探究
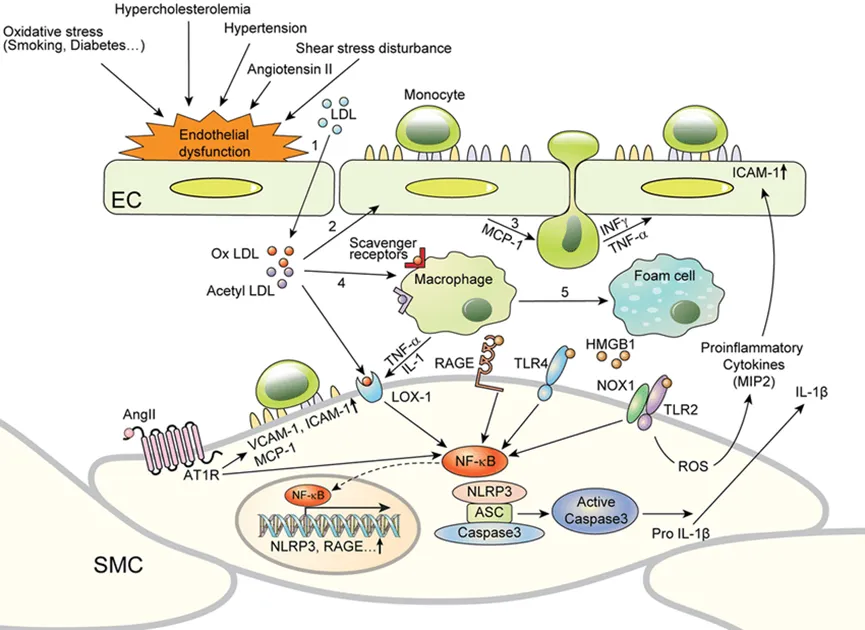
低密度脂蛋白(LDL)通过内皮层转运至内皮下区域,并在那里经历氧化过程,这一事件标志着动脉粥样硬化的起始。最新的单细胞转录组学研究揭示,内皮细胞是主动脉构成中的主要细胞类型之一。CD36广泛分布于整个主动脉内皮细胞的表面。在模拟氧化低密度脂蛋白(ox-LDL)暴露或高脂饮食条件下,CD36的表达水平上升,这加速了ox-LDL或其他改性脂质在主动脉中的摄取。相比之下,在CD36基因敲除(CD36-/-)的小鼠中,由CD36介导的ox-LDL摄取能力减弱。CD36促进的LDL内吞作用在大鼠主动脉或冠状动脉中,是动脉粥样硬化发病机制的关键环节。利用内皮细胞特异性CD36基因敲除的小鼠模型发现,内皮细胞内长链脂肪酸的转运减少,糖耐量得到改善,且心脏脂质沉积降低。这可能是由于CD36介导的ox-LDL摄取依赖于脂肪酸(FAs),CD36通过提升细胞内酯化速率来促进FAs的吸收。因此,FAs与CD36的结合增强了CD36介导的ox-LDL摄取,这一连串反应最终促进了动脉粥样硬化的发生。
探究CD36如何影响血管平滑肌细胞的功能障碍
血管平滑肌细胞(VSMCs)作为动脉壁的关键组成部分,对维持血管的正常张力起着至关重要的作用。当VSMCs的行为、功能及其抗氧化状态发生异常变化时,会加速动脉粥样硬化斑块的进展。值得注意的是,CD36在VSMCs表面有所表达,并与氧化低密度脂蛋白(ox-LDL)诱导的脂质代谢紊乱以及动脉壁免疫应答失衡紧密相关。在先天免疫系统中,CD36扮演着模式识别受体的角色。
研究表明,在原代培养的VSMCs中,ox-LDL能够诱导粒细胞集落刺激因子(G-CSF)和粒细胞-巨噬细胞集落刺激因子(GM-CSF)的表达增强,并提升细胞的迁移能力,而这些效应可以被CD36特异性的小干扰RNA(siRNA)所逆转。此外,VSMCs的增殖以及从中膜向内膜的迁移,会导致动脉壁增厚(即新内膜形成),这是血栓形成的一个重要风险因素。
从CD36基因敲除(CD36-/-)小鼠的胸主动脉中分离的VSMCs,其增殖能力受到显著抑制。这种增殖抑制的表型可能与细胞周期蛋白A的表达下调有关。因此,针对血管系统中的CD36进行干预,为治疗血管损伤和其他血管相关疾病提供了新的潜在靶点。
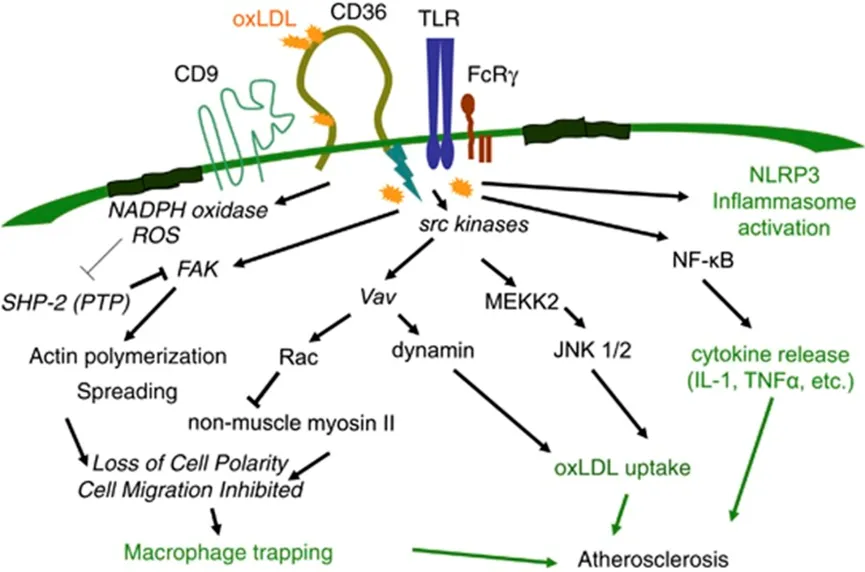
CD36对巨噬细胞功能异常的关键影响
在血脂异常条件下,低密度脂蛋白(LDL)颗粒会在内皮下空间积累并发生氧化修饰,这一过程吸引着单核细胞迁移并分化。氧化型低密度脂蛋白(ox-LDL)能够通过过氧化物酶体增殖物激活受体γ(PPARγ)诱导CD36的表达,从而促进巨噬细胞对ox-LDL的摄取,使巨噬细胞转变为富含脂质的泡沫细胞。细胞内活性氧(ROS)是CD36促进动脉粥样硬化作用的主要因素之一,尽管抑制NADPH氧化酶并未完全阻断CD36的这一作用,暗示还有其他关键调节因子参与其中。线粒体作为ROS的主要来源,可能是ox-LDL/CD36轴的下游效应器,并通过线粒体途径调节ROS的产生和炎症状态。此外,CD36还参与了巨噬细胞代谢的重编程,CD36缺失的巨噬细胞在ox-LDL处理后表现出氧化磷酸化增强,呈现M1巨噬细胞表型,而野生型巨噬细胞则转向糖酵解。
巨噬细胞迁移能力的受损会导致其滞留在内皮下间隙。ox-LDL能够抑制CD36缺陷细胞的迁移能力和肌动蛋白聚合,而这一过程在CD36缺陷细胞中被阻断。CD36通过NADPH氧化依赖性ROS生成来调节巨噬细胞的迁移,影响SHP2和FAK的活化。使用CD36配体EP80317治疗可以减少ApoE缺失小鼠中巨噬细胞向动脉粥样硬化病变的招募,但在CD36缺失小鼠中这一作用被阻断,表明该配体可能通过竞争性地减少ox-LDL与CD36的结合来发挥作用。
此外,CD36还参与调节巨噬细胞从M1向M2极化的过程,这是增强巨噬细胞迁移能力的关键步骤。M2巨噬细胞的激活需要脂肪酸(FA)氧化来支持,而CD36占据甘油三酯底物产生足够的FA,从而促进极性的丧失。睾丸孤核受体4(TR4)能够感知FA的增加,进而促进CD36的表达,形成恶性循环,最终促进泡沫细胞的形成。最近的研究还发现,CD36参与ox-LDL诱导的巨噬细胞中波形蛋白(vimentin)的分泌以及促炎细胞因子(如TNF-α和IL-6)的释放。这些发现表明,抑制CD36可以防止巨噬细胞极化、迁移,并有助于预防动脉粥样硬化。
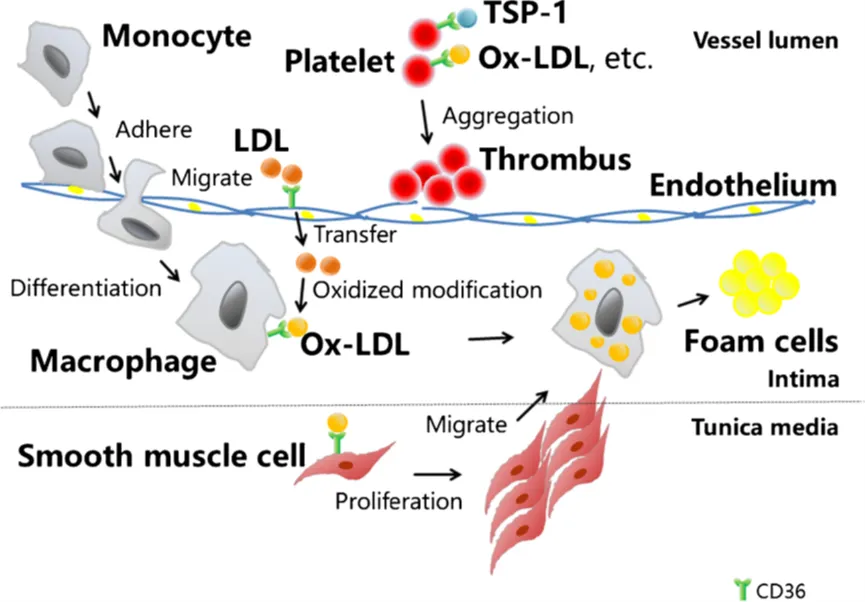
CD36和血小板活化
慢性血管损伤是一个持续存在但往往隐匿的过程,直至在内皮和纤维帽中发展成高度炎症的斑块,并最终发生破裂。这一急性损伤事件触发血小板的募集和附着于受损内皮表面,随后配位分子的动员导致血小板聚集,最终形成纤维蛋白凝块和血栓。深入探究血小板促血栓形成表型的机制,有望为血栓相关疾病的治疗提供新的靶点。
多种内源性或外源性分子,如氧化型低密度脂蛋白(ox-LDL),能够与血小板表面的CD36结合,通过一系列信号传导途径,包括Src家族激酶、Vav-鸟嘌呤核苷酸交换因子、cGMP以及NADPH氧化酶,驱动血小板的高度激活状态。此外,晚期糖基化终产物也能直接与血小板衍生的CD36相互作用,加速血栓的形成过程,而在CD36基因敲除(CD36-/-)的小鼠中,闭塞性血栓的形成时间明显延迟。
血小板上的CD36不仅通过ERK5途径增强体内caspase依赖的促凝活性,还在ox-LDL的刺激下促进血小板与单核细胞的聚集,这种聚集反过来又增强单核细胞以CD36依赖的方式摄取ox-LDL。值得注意的是,冠状动脉疾病患者的血小板中CD36的表达水平显著高于无病人群。更重要的是,这些患者的血小板与单核细胞共培养时,能够诱导单核细胞的极化和泡沫细胞的形成,进一步加剧了动脉粥样硬化的进程。
综上所述,血小板表面的CD36在血栓形成和动脉粥样硬化的发展中扮演了重要角色,因此,针对CD36的干预策略可能为血栓相关疾病的治疗提供新的途径。
结语
CD36作为一种多功能的炎症代谢受体,在生理条件下发挥着调节脂质稳态和免疫稳态的重要作用。然而,CD36表达的失衡与动脉粥样硬化的发生和发展密切相关。在这一过程中,不同CD36表达细胞之间的复杂相互作用起到了关键的推动作用。
展望未来,CD36相关领域的研究将向多个方向深入发展。首先,新型长链非编码RNA对CD36的转录后调控机制及其对血管功能障碍的影响将成为研究的热点。其次,组蛋白修饰,如乙酰化和甲基化,对CD36表达的调控作用也将受到更多的关注。此外,CD36单核苷酸多态性在动脉粥样硬化中的致病作用,以及CD36的可溶性形式作为动脉粥样硬化诊断的潜在可靠生物标志物,都是值得深入探索的重要方向。
综上所述,对CD36及其相关机制的深入研究,不仅有助于揭示动脉粥样硬化的发病机理,还可能为相关疾病的预防和治疗提供新的思路和策略。
| 名称 | 货号 | 规格 |
| FITC Mouse Anti-Human CD36(CB38) | 561820 | 25Tst |
| APC Mouse Anti-Human CD36(CB38) | 561822 | 25Tst |
| CD36 Recombinant Rabbit mAb (SDT-358-19) | S0B2216-1ml | 1ml |
| CD36 (D8L9T) Rabbit mAb | 14347S | 100ul |